Parallelize GEOS-Chem and HEMCO source code
Single-node paralellization in GEOS-Chem Classic and HEMCO is acheieved with OpenMP. OpenMP directives, which are included in every modern compiler, allow you to divide the work done in DO loops among several computational cores. In this Guide, you will learn more about how GEOS-Chem Classic and HEMCO utilize OpenMP.
Overview of OpenMP parallelization
Most GEOS-Chem and HEMCO arrays represent quantities on a geospatial grid (such as meteorological fields, species concentrations, production and loss rates, etc.). When we parallelize the GEOS-Chem and HEMCO source code, we give each computational core its own region of the “world” to work on, so to speak. However, all cores can see the entire “world” (i.e. the entire memory on the machine) at once, but is just restricted to working on its own region of the “world”.
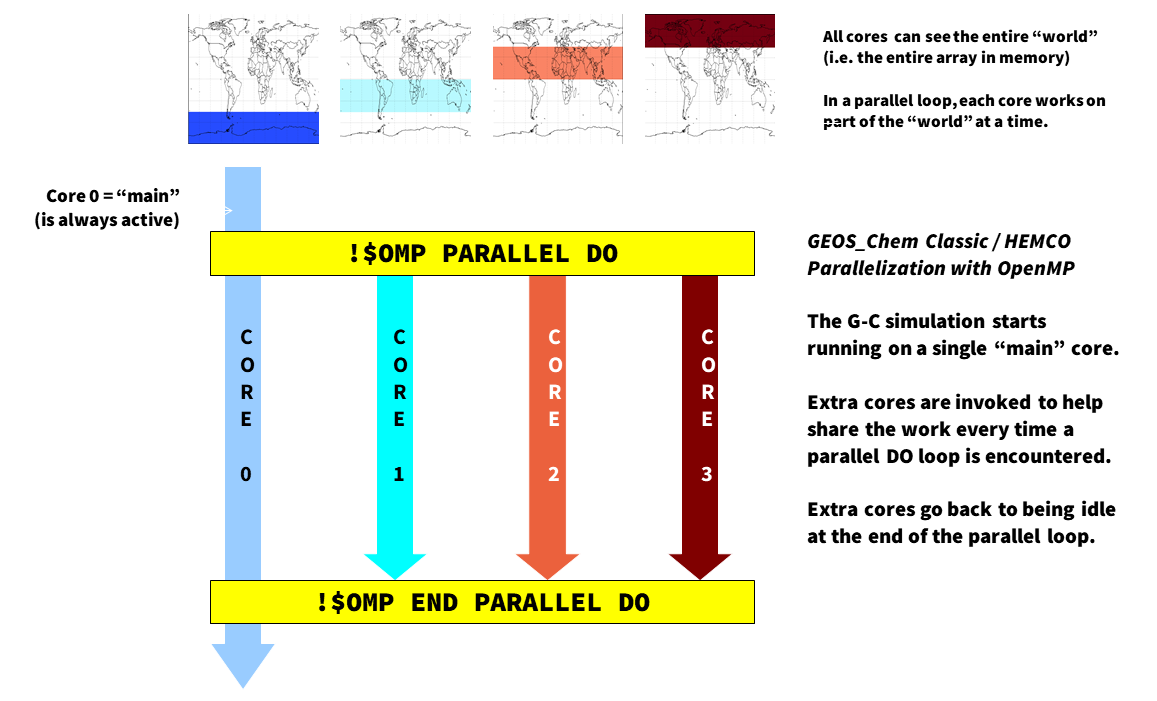
It is important to remember that OpenMP is loop-level parallelization. That means that only commands within selected DO loops will execute in parallel. GEOS-Chem Classic and HEMCO (when running within GEOS-Chem Classic, or as the HEMCO standalone) start off on a single core (known as the “main core”). Upon entering a parallel DO loop, other cores will be invoked to share the workload within the loop. At the end of the parallel DO loop, the other cores return to standby status and the execution continues only on the “main” core.
One restriction of using OpenMP parallelization is that simulations may use only as many cores that share by the same memory. In practice, this limits GEOS-Chem Classic and HEMCO standalone simulations to using 1 node (typically less than 64 cores) of a shared computer cluster.
We should also note that GEOS-Chem High Performance (aka GCHP) uses a different type of parallelization (MPI). This allows GCHP to use hundreds or thousands of cores across several nodes of a computer cluster. We encourage you to consider using GCHP for hour high-resolution simulations.
Example using OpenMP directives
Consider the following nested loop that has been parallelized with OpenMP directives:
!$OMP PARALLEL DO &
!$OMP SHARED( A ) &
!$OMP PRIVATE( I, J, B ) &
!$OMP COLLAPSE( 2 ) &
!$OMP SCHEDULE( DYNAMIC, 4 )
DO J = 1, NY
DO I = 1, NX
B = A(I,J)
A(I,J) = B * 2.0
ENDDO
ENDDO
!$OMP END PARALLEL DO
This loop will assign different (I,J) pairs to different
computational cores. The more cores specified, the less time it will
take to do the operation.
Let us know look at the important features of this loop.
- !$OMP PARALLEL DO
This is known as a loop sentinel. It tells the compiler that the following DO-loop is to be executed in parallel. The clauses following the sentinel specify further options for the parallelization. These clauses may be spread across multiple lines by using a continuation command (
&) at the end of the line.
- !$OMP SHARED( A )
This clause tells the compiler that all computational cores can write to
Asimultaneously. This is OK because each core will recieve a unique set of(I,J)pairs. Thus data corruption of theAarray will not happen. We say thatAis a SHARED variable.Note
We recommend using the clause
!$OMP DEFAULT( SHARED ), which will declare all varaiables as shared, unless they are explicitly placed in an!$OMP PRIVATEclause.
- !$OMP PRIVATE( I, J, B )
Because different cores will be handling different
(I,J)pairs, each core needs its own private copy of variablesIandJ. The compiler creates these temporary copies of these variables in memory “under the hood”.If the
IandJvariables were not declaredPRIVATE, then all of the computational cores could simultaneously write toIandJ. This would lead to data corruption. For the same reason, we must also place the variableBwithin the!$OMP PRIVATEclause.
- !$OMP COLLAPSE( 2 )
By default, OpenMP will parallelize the outer loop in a set of nested loops. To gain more efficiency, we can vectorize the loop. “Under the hood”, the compiler can convert the two nested loops over
NXandNYinto a single loop of sizeNX * NY, and then parallelize over the single loop. Because we wish to collapse 2 loops together, we use the!$OMP COLLAPSE( 2 )statement.
- !$OMP SCHEDULE( DYNAMIC, 4 )
Normally, OpenMP will evenly split the domain to be parallelized (i.e.
(NX, NY)) evenly between the cores. But if some computations take longer than others (i.e. photochemistry at the day/night boundary), this static scheduling may be inefficient.The
SCHEDULE( DYNAMIC, 4 )will send groups of 4 grid boxes to each core. As soon as a core finishes its work, it will immediately receive another group of 4 grid boxes. This can help to achieve better load balancing.
- !$OMP END PARALLEL DO
This is a sentinel that declares the end of the parallel DO loop. It may be omitted. But we encourage you to include them, as defining both the beginning and end of a parallel loop is good programming style.
Environment variable settings for OpenMP
Please see Set environment variables for parallelization to learn which environment variables you must add to your login environment to control OpenMP parallelization.
OpenMP parallelization FAQ
Here are some frequently asked questions about parallelizing GEOS-Chem and HEMCO code with OpenMP:
How can I tell what should go into the !$OMP PRIVATE clause?
Here is a good rule of thumb:
All variables that appear on the left side of an equals sign, and that have lower dimensionality than the dimensionality of the parallel loop must be placed in the
!$OMP PRIVATEclause.
In the example shown above, I,
J, and B are scalars, so their dimensionality
is 0. But the parallelization occurs over two DO loops (1..NY
and 1..NX), so the dimensionality of the parallelization is 2.
Thus I, J, and B must go inside the
!$OMP PRIVATE clause.
Tip
You can also think of dimensionality as the number of indices a
variable has. For example A has dimensionality 0, but
A(I) has dimensionality 1, A(I,J) has
dimensionality 2, etc.
Why do the !$OMP statements begin with a comment character?
This is by design. In order to invoke the parallel procesing commands,
you must use a specific compiler command (such as -openmp,
-fopenmp, or similar, depending on the compiler). If you
omit these compiler switches, then the parallel processing directives
will be considered as Fortran comments, and the associated DO-loops
will be executed on a single core.
Do subroutine variables have to be declared PRIVATE?
Consider this subroutine:
SUBROUTINE mySub( X, Y, Z )
! Dummy variables for input
REAL, INTENT(IN) :: X, Y
! Dummy variable for output
REAL, INTENT(OUT) :: Z
! Add X + Y to make Z
Z = X + Y
END SUBROUTINE mySub
which is called from within a parallel loop:
INTEGER :: N
REAL :: A, B, C
!$OMP PARALLEL DO &
!$OMP DEFAULT( SHARED ) &
!$OMP PRIVATE( N, A, B, C )
DO N = 1, nIterations
! Get inputs from some array
A = Input(N,1)
B = Input(N,2)
! Add A + B to make C
CALL mySub( A, B, C )
! Save the output in an array
Output(N) = C
ENDDO
!$OMP END PARALLEL DO
Using the rule of thumb described above, because N, A,
B, and C are scalars (having dimensionality = 0), they
must be placed in the !$OMP PRIVATE clause.
But note that the variables X, Y, and Z do not
need to be placed within a !$OMP PRIVATE clause within
subroutine mySub. This is because each core calls
mySub in a separate thread of execution, and will create its
own private copy of X, Y, and Z in memory.
What does the THREADPRIVATE statement do?
Let’s modify the above example
slightly. Let’s now suppose that subroutine mySub from the prior
example is now part of a Fortran-90 module, which looks like this:
MODULE myModule
! Module variable:
REAL, PUBLIC :: Z
CONTAINS
SUBROUTINE mySub( X, Y )
! Dummy variables for input
REAL, INTENT(IN) :: X, Y
! Add X + Y to make Z
! NOTE that Z is now a global variable
Z = X + Y
END SUBROUTINE mySub
END MODULE myModule
Note that Z is now a global scalar variable with
dimensionality = 0. Let’s now use the same parallel loop
(dimensionality = 1) as before:
! Get the Z variable from myModule
USE myModule, ONLY : Z
INTEGER :: N
REAL :: A, B, C
!$OMP PARALLEL DO &
!$OMP DEFAULT( SHARED ) &
!$OMP PRIVATE( N, A, B, C )
DO N = 1, nIterations
! Get inputs from some array
A = Input(N,1)
B = Input(N,2)
! Add A + B to make C
CALL mySub( A, B )
! Save the output in an array
Output(N) = Z
ENDDO
!$OMP END PARALLEL DO
Because Z is now a global variable with lower dimensionality
than the loop, we must try to place it within an !$OMP PRIVATE
clause. However, Z is defined in a different program unit
than where the parallel loop occurs, so we cannot place it in an
!$OMP PRIVATE clause for the loop.
In this case we must place Z into an !$OMP
THREADPRIVATE clause within the module where it is declared, as shown
below:
MODULE myModule
! Module variable:
! This is global and acts as if it were in a F77-style common block
REAL, PUBLIC :: Z
!$OMP THREADPRIVATE( Z )
... etc ...
This tells the computer to create a separate private copy of Z
in memory for each core.
Important
When you place a variable into an !$OMP PRIVATE or
!$OMP THREADPRIVATE clause, this means that the variable
will have no meaning outside of the parallel loop where it is used.
So you should not rely on using the value of PRIVATE or
THREADPRIVATE variables elsewhere in your code.
Most of the time you won’t have to use the !$OMP THREADPRIVATE
statement. You may need to use it if you are trying to parallelize code
that came from someone else.
Can I use pointers within an OpenMP parallel loop?
You may use pointer-based variables (including derived-type objects) within an OpenMP parallel loop. But you must make sure that you point to the target within the parallel loop section AND that you also nullify the pointer within the parallel loop section. For example:
INCORRECT:
! Declare variables
REAL, TARGET :: myArray(NX,NY)
REAL, POINTER :: myPtr (: )
! Declare an OpenMP parallel loop
!$OMP PARALLEL DO ) &
!$OMP DEFAULT( SHARED ) &
!$OMP PRIVATE( I, J, myPtr, ...etc... )
DO J = 1, NY
DO I = 1, NX
! Point to a variable.
!This must be done in the parallel loop section.
myPtr => myArray(:,J)
. . . do other stuff . . .
ENDDO
!$OMP END PARALLEL DO
! Nullify the pointer.
! NOTE: This is incorrect because we nullify the pointer outside of the loop.
myPtr => NULL()
CORRECT:
! Declare variables
REAL, TARGET :: myArray(NX,NY)
REAL, POINTER :: myPtr (: )
! Declare an OpenMP parallel loop
!$OMP PARALLEL DO ) &
!$OMP DEFAULT( SHARED ) &
!$OMP PRIVATE( I, J, myPtr, ...etc... )
DO J = 1, NY
DO I = 1, NX
! Point to a variable.
!This must be done in the parallel loop section.
myPtr => myArray(:,J)
. . . do other stuff . . .
! Nullify the pointer within the parallel loop
myPtr => NULL()
ENDDO
!$OMP END PARALLEL DO
In other words, pointers used in OpenMP parallel loops only have meaning within the parallel loop.
How many cores may I use for GEOS-Chem or HEMCO?
You can use as many computational cores as there are on a single node of your cluster. With OpenMP parallelization, the restriction is that all of the cores have to see all the memory on the machine (or node of a larger machine). So if you have 32 cores on a single node, you can use them. We have shown that run times will continue to decrease (albeit asymptotically) when you increase the number of cores.
Why is GEOS-Chem is not using all the cores I requested?
The number of threads for an OpenMP simulation is determined by the
environment variable OMP_NUM_THREADS.
You must define OMP_NUM_THREADS in your environment file
to specify the desired number of computational cores for your
simulation. For the bash shell, use4 this command to
request 8 cores:
export OMP_NUM_THREADS=8
MPI parallelization
The OpenMP parallelization used by GEOS-Chem Classic and HEMCO standalone is an example of shared memory parallelization (also known as serial parallelization). As we have seen, we are restricted to using a single node of a computer cluster. This is because all of the cores need to talk with all of the memory on the node.
On the other hand, MPI (Message Passing Interface) parallelzation is an example of distributed parallelization. An MPI library installation is required for passing memory from one physical system to another (i.e. across nodes).
GEOS-Chem High Performance (GCHP) uses Earth System Model Framework (ESMF) and MAPL libraries to implement MPI parallelization. For detailed information, please see gchp.readthedocs.io.